For many of projects I need specialized code that I could not find anywhere else. In the (faint) hope that my efforts might be useful to others as well and to allow anyone to reproduce my own results I make my software available here.
Release source code archives are found on this page. The source code history and current development version for most packages can be accessed directly via my git repositories at github.com/orbeckst. Please use the Issue Tracker associated with a package for feature requests and bug reports. Thanks!
The software accessible through this page is Copyright © 2001-2010 Oliver Beckstein and is made available under the terms of the GNU General Public License, unless otherwise noted in the package. Please see the files README and INSTALL which accompany each package, or the comments at the top of the files. If you require different licensing terms please contact me.

Hop is a python package based on MDAnalysis to analyze solvation dynamics as outlined at Biophysics 2009 in the poster presentation Quantitative Analysis of Water Dynamics in and near Proteins, Oliver Beckstein, Naveen Michaud-Agrawal and Thomas B. Woolf, doi:10.1016/j.bpj.2008.12.3147.
The package will be made available here and on github.com/orbeckst/hop. Inquiries for beta-testing can be directed to me. Beta-testers can file bugs and requests for enhancements through the Issue Tracker.
lipidbook.bioch.ox.ac.uk is a public repository for force field parameters with a special emphasis on lipids, detergents, and similar molecules that are of interest when simulating biological membranes. It acts as a warehouse for parameter files that are supplied by the community.
Lipidbook is described in

It is hosted at mdanalysis.googlecode.com, including Online Documentation, a source code repository, and the MDAnalysis Issue Tracker.
The paper on MDAnalysis explains the philosophy and general layout of the toolkit together with a number of examples:
Note that the Python code in the online html version of the paper is useless because the crucial indentation of Python was not preserved in the conversion process. The PDF version is mostly correct although the final formatted version ended up with a few line breaks that are illegal in the Python syntax. The free PubmedCentral version (available April 2012), however, contains perfectly formatted Python code.
Corrected code is listed here (and on the MDAnalysis Paper Errata page) and can also be found in the examples that are part of the MDAnalysis package. If you find further problems then please let us know by filing an issue in the MDAnalysis Issue Tracker or contacting me by email.
universe.selectAtoms("byres (resname SOL and around 3.5 protein)").write("solvation-shell.pdb")
def blocked(universe, nblocks, analyze):
size = universe.trajectory.numframes/nblocks
blocks = []
for block in xrange(nblocks):
a = []
for ts in u.trajectory[block*size:(block+1)*size]:
a.append(analyze(universe))
blocks.append(numpy.average(a))
blockaverage = numpy.average(blocks)
blockstd = numpy.std(blocks)
return nblocks, size, blockaverage, blockstd
Radius of gyration analysis function
def rgyr(universe):
return universe.selectAtoms("protein").radiusOfGyration()
Time series analysis
MDAnalysis.collection.addTimeseries(\
Timeseries.Dihedral(atomselection))
data = universe.trajectory.correl(\
MDAnalysis.collection, skip=10)
examples/radial_distribution_function.py
from the distribution.A source code repository is hosted at github.com/orbeckst/GromacsWrapper, which also contains the GromacsWrapper Issue Tracker for reports of bugs and feature requests.

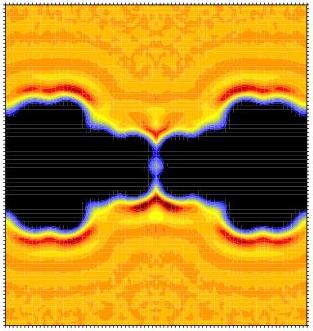
gridcount is an analysis tool for Gromacs that creates 3D (number)
densities from molecular dynamics trajectories. Typically, this is
used to look at the density of water or ions near proteins or in
channels and pores (left: water density in a model pore,
contoured at 0.01 of the bulk density, and right radially
averaged density). The package consists of two programs,
g_ri3Dc and a_ri3Dc.
g_ri3Dc counts occurrences of molecules on a 3D grid over
a MD trajectory. A typical application is to choose the water index
group as the molecules to be counted. This creates (after proper
normalisation) a 3D density map. The map is written in a
system-independent (xdr) grid file for once and all.
The grid file is read in by a_ri3Dc, a simple
analysis and converter tool. At the moment it is mainly useful to
calculate cylindrical and slice averages and convert the density map
into gOpenMol's plt
format, which can also be read by VMD (from version
1.8.2 onwards). Alternatively, it also outputs the whole grid in ASCII
format. 1D graphs can be displayed with xmgrace and for 2D
data, parameter and data files are written, which are suitable for xfarbe.
a_gridcalc (in version 1.1) can average densities generated with
g_ri3Dc (and provides framework code for further grid
manipulations).
If you want to access densities in Python then you can install my
GridDataFormats
package, which can read the gOpenMol plt files and makes data
available as NumPy arrays. (GridDataFormats can also be found on PyPi/GridDataFormats so you can simply install it with easy_install
GridDataFormats or pip install
GridDataFormats.)
The source code repository is at github.com/orbeckst/gridcount. You can download the latest development version from there, too. Please file bug reports and feature requests through the gridcount Issue Tracker.
When you use gridcount please cite
git clone -b gromacs_4.5 https://github.com/orbeckst/gridcount.gitAlternatively, download a tarball of the repository snapshot: gridcount-gmx4.5-1.4.tar.gz.
Makefile but you can also run make
check to check if the Gromacs libraries and include files have
been found.dprintf) that showed up on
newer Linux versions (in utilgmx.h and
/usr/include/stdio.h)
g_count and g_flux are simple Gromacs tools
that are primarily useful to characterize movement of water inside a
cylindrical region. This can be the pore of an ion channel or a carbon
nanotube. g_count returns statistics about how many atoms
of a certain type (for instance, water oxygens) occupied the region of
interest. g_flux calculates the flux through a cylindrical
region. g_zcoord is a dumb piece of code that just prints
z-coordinates of atoms in a cylindrical region; it should be
easy enough to add additional functionality.
g_flux is more sophisticated than g_count
and gives more detailed information but you are encouraged to check
that the reported results agree with what you can see in your
system. For instance, get the indices of translocating particles and
then track them in a trajectory viewer such as VMD.
Detailed documentation can be found in the help functions for each
program (run it with -h). Background information
(slighly outdated for the current g_count-gmx4.5 release) can be found in the
appendix
(pdf) of
my thesis.
A source code repository is hosted at github.com/orbeckst/g_count, which also contains the g_count Issue Tracker for reports of bugs and feature requests.
When you use g_flux/g_count please cite
make check to
catch problems with missing include files and libraries
before compilingdprintf (patch contributed
by Jayne E Wallace)g_flux and Gromacs 4.0.4 vs
4.0.2. It appears that some waters are counted that are not
really transitioning when g_flux was compiled
against 4.0.4. Please check your results carefully and please
send feedback as I am trying to find out what is going on. This
probably affects all gmx4.x versions.-sojourns flag to write everything, i.e. transitions
and sojourns.
A transition is a true crossing event when the particle
enters on one side and exits on the other. A sojourn is
logged when a particle enters and exits on the same side. Note
that logged sojourns are only meaningful if they have a starting
time t > 0 because of the way the crossing detection
algorithm is implemented.g_flux: records whenever a particle spent time in
the cavity; includes both successful transitions and sojourns.
g_flux option
-ncr) had residue numbers and not atom numbers;
thus it could not be used as a normal index file.
g_zcoord and removed cruft; otherwise
like gmx4.2.g_flux should now count particles only inside
the cylinder; this did not work before but all options to this effect
were silently ignored. Tyically only matters for systems that are not
embedded in an impenetrable membrane.g_zcoord not supported; use the standard g_traj
-nox -noy -ox coord.xvg for the basic functionality.gmxGromacsMajorVersion.g_countVersion; this should show immediately which version of Gromacs is required for compilation.
g_flux silently ignores the -R
radius option.Earlier versions of g_count are functionally identical to g_count-0.3 but compile against earlier versions of Gromacs. See the README.
g_flux
acts as if the cylinder had an infinite radius).
gmx_rescue is a small program, which
originated on the gmx-l mailing list. It was cleaned up by Marc Baaden. It allows one to
read accross errors in xtc trajectory files, eg when gmxcheck
complains Fatal error: Magic Number Error in XTC file (read 0,
should be 1995) or similarly, Reading frame ... Floating
point exception. gmx_rescue64 is a modified
version, which can cope with large files on Linux (> 2GB). The
strategy to salvage the good frames is simple:
trjconv.gmx_rescue64 to get all the frames
after the broken ones.gmxcheck
the new fragment. If more errors come up, repeat with 1.trjcat all xtc segments that you accumulated.Note, that this will leave holes in your xtc where your atoms will appear to jump. That might or might not severly influence your subsequent analysis.
gmx_recue64), or the whole package gmx_rescue.tgz
from Marc Baaden's
download page, or from the Gromacs contributions
page (the gmx_rescue_LFS package, but it might
contain an older version of gmx_rescue64). Parts of gmx_rescue is © 2003 Marc Baaden; parts
of gmx_rescue64 is © 2003 Marc Baaden, 2004 Oliver
Beckstein.
Apply this patch to Alan Grossfield's wham code so that it can be used with the pdo.gz files that are output by Gromacs' umbrella sampling code. There are two versions of the patch. The older one works for Alan's 1.x code base, the newer one is based on his 2.0.2 release. Note that my patch only applies to the 1D-wham code; the 2D-wham is unchanged.
Download the appropriate patch and do the following (for the new 2.0.2 wham)
wget http://membrane.urmc.rochester.edu/wham/wham-release-2.0.2.tgz mkdir wham_gmx cd wham_gmx tar -zxvf ../wham-release-2.0.2.tgz bunzip2 -c ../gromacsified_wham_2.0.2-ob1.diff.bz2 | patch -p0This should apply cleanly without problems (just make sure to execute the patch command in the directory in which the directories `wham', `wham-2d', `nr' , `doc' appear). Then compile the 1d-wham that can read pdo.gz files:
cd wham
make
make PREFIX=${HOME} install
The last line will install the wham binary under
~/bin/wham. Just running wham without
arguments will give you usage information.
wham requires a so-called meta data file as
input. The pdo2meta.sh script in the
scripts/ directory automatically creates this file from a
list of pdo (or pdo.gz) files. There's also a program for testing the
code in extras/fakepmf; see wham/README.txt and the
source code for details.
easy_install -f http://sbcb.bioch.ox.ac.uk/users/oliver/download/Python
PACKAGE_NAME
Typically, these packages are also hosted on PyPI and therefore a simple
easy_install PACKAGE_NAME
should be tried first.
A number of bash functions and shell or perl scripts that I use and
haven't seen somewhere else (but then again, I didn't look too
hard...). Feel free to take what you need but remember that it DOES
NOT COME WITH ANY WARRANTY, NEITHER EXPLICITLY NOR IMPLICITLY
IMPLIED. Documentation is provided through standard usage notes (try
-h or --help) and of course through, erm,
self-documenting source.
Direct access to my Gromacs code download directory; maybe you find something useful. Good luck!
