Research
Overview
The research interests of the group are broadly centred on protein dynamics and drug design. We use and develop a variety of computational tools to improve our understanding of how ligand-binding controls conformational change in proteins. A better understanding of this should enable more rational design of drugs and treatments. Thus much of our work is in collaboration with pharmaceutical companies. We have good local computational resources, but we also make extensive use of ARCHER via the HECBioSim Consortium. We are also founder members of the CCPBioSim - a collaborative computational project for Biomolecular simulation. The following sections provide a little more detail on the areas of current research.
Synaptic Receptors
We have had a long-standing interest in developing and applying computational methods including docking and molecular dynamics simulations to receptor proteins such as the ligand-gated ion channels. These are receptors that upon binding of a ligand change their conformation such that ions can pass through a central pore and down their electrochemical gradient. We are particularly interested in two distinct families of these receptors: 1. The ionotropic glutamate receptors (shown below) and 2. The nicotinic acetylcholine receptor. Although there has been a recent increase in the amount of structural information available, many questions still remain concerning the dynamics associated with their function, not least of which is how does the binding of a ligand lead to an opening of the channel? Aside from this question we have been asking what controls activation and desensitization. In the case of kainate receptors, (a subtype of glutamate receptor) there is a distinct role for cations and anions [1,2].
Figure 1: Structure of an ionotropic glutamate receptor. How does binding of glutamate lead to opening of the transmembrane pore? |
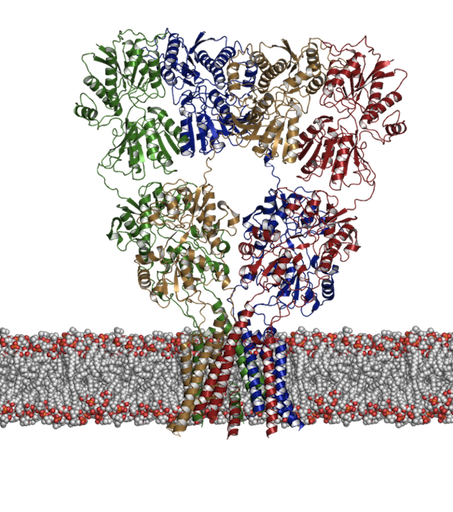 |
Conformational Dynamics
One of our more fundamental areas of research is focussed on developing and utilizing tools to compare the dynamics of different proteins. Although the relationship between sequence and structure is well-defined and various methods exist to calculate the similarities at both levels, the assessment of the similarity of the dynamics presents more challenges – literally a moving target. Our research has shown how the dynamics of proteins (form molecular dynamics simulations) can be compared via different methods [3-5].
As well as providing a fundamental level of understanding we use the tools we develop here to guide our interpretation of biochemically interesting proteins, and in particular transporter proteins. Two examples currently under investigation in the lab are the P-glycoprotein (P-gP) [6,7] (illustrated below) and the SV2A protein, the latter is believed to be similar in structure to the MFS family of transporter proteins.
Figure 2: Structure of P-glycoprotein embedded in a lipid bilayer. P-gp is comprised of two pseudosymmetric domains (colour red and blue) which bind a variety of different drugs in the central cavity. ATP binding and hydrolysis leads to expulsion of the drug to the extracellular (top in the is diagram) side of the membrane |
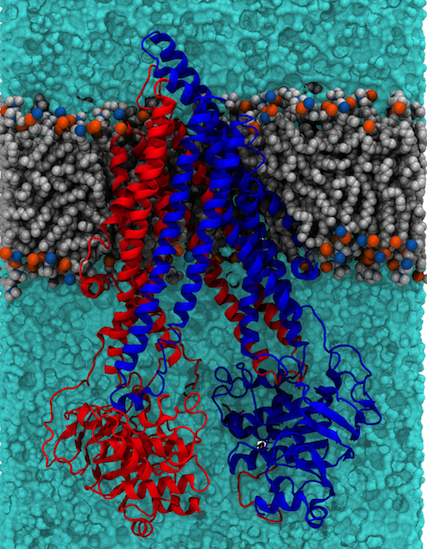 |
GPCRs
Structural biology of G-Protein Coupled Receptors has made incredible advances in the last few years with several atomic-resolution structures now available. We are working in partnership with Evotec to use molecular dynamics to help refine models of GPCR drug targets to help rationalize drug design. One family of GPCRs that we have been working on is the Orexin family. In particular we were able to show how MD-refined models of both the Orexin-1 and Orexin-2 receptors can be used to rationalize not only all of the extensive QSAR data which has been published to date, but also the underlying basis of selectivity in these two similar receptors. [8] The figure below shows a top down view of the receptors with the dual antagonist almorexant and illustrates the key difference between the OX1 and OX2 receptors in terms of transmembrane helix III.
| Figure 3: Top down view comparing the OX1 and OX2 receptors with the dual antagonist almorexant. The key difference is found in TM3 for the OX2 receptor (pink) where it is displaced outward compared to the OX1 receptor (cyan). | 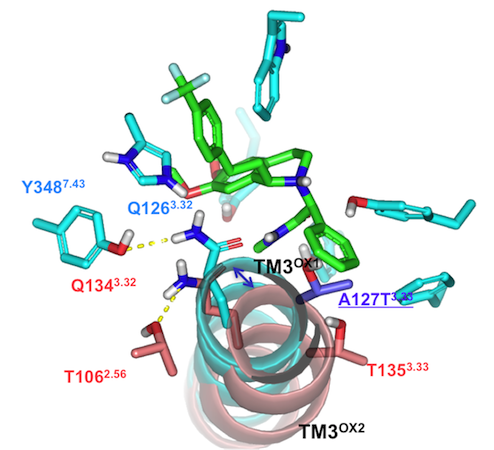 |
Drug-Design
We are also interested in the more fundamental aspects of drug design. In recent years there has been a lot of discussion about the role of water in drug design. In particular, it has been suggested that by knowing where water molecules bind and being able to predict whether they would be displaceable or not could be used to design compounds that utilize or displace water molecules that are found within binding pockets. One way to predict this is to perform rigorous free energy calculations. However, that is still way too time-consuming to be a realistic tool in high throughput virtual screening. We have developed an approach that is able to predict the positions of water molecules in binding sites and classify them according to whether they will be displaceable or not [9]. More information about that is available from the WaterDock webpage.
Related to this, in the world of virtual screening, scoring functions are often used to provide an indication of how well a compound will bind to a particular protein. We have been assessing (under review) what the maximum error one can expect to see from that type of approach. The results suggest that there is no such thing as a “universal scoring function” and that theoretically you will always be better off using a target-specific scoring function. Most people in the field have recognized this anecdotally, but until now, there was no formal proof.
References
1. Vijayan R, Plested AJR, Mayer ML, Biggin PC (2009) Selectivity and cooperativity of modulatory ions in a neurotransmitter receptor. Biophysical Journal 96: 1751-1760.2. Plested AJR, Vijayan R, Biggin PC, Mayer ML (2008) Molecular basis of Kainate receptor modulation by sodium. Neuron 58: 720-735.
3. Münz M, Hein J, Biggin PC (2012) The role of flexibility and conformational selection in the binding promiscuity of PDZ domains. PLoS Comput Biol 8: e1002749.
4. Münz M, Lyngsø R, Hein J, Biggin PC (2010) Dynamics-based alignment of proteins: An alternative approach to quantify dynamic similarity. BMC Bioinformatics 11: 118.
5. Pang A, Arinaminpathy Y, Sansom MSP, Biggin PC (2005) Comparative molecular dynamics - Similar folds and similar motions? Proteins: Structure, Function and Genetics 61: 809-822.
6. Marcoux J, Wang SC, Politis A, Reading E, Ma J, et al. (2013) Mass spectrometry reveals synergistic effects of nucleotides, lipids, and drugs binding to a multidrug resistance efflux pump. Proc Natl Acad Sci USA epub online.
7. Ma J, Biggin PC (2013) Substrate versus inhibitor dynamics of P-glycoprotein. Prot Struc Func Gen epub online.
8. Heifetz A, Morris GB, Biggin PC, Barker O, Fryatt T, et al. (2012) Study of human Orexin-1 and -2 -G-protein-coupled receptors with novel and published antagonists by modeling, molecular dynamics simulations, and site-directed mutagenesis. Biochemistry 51: 3178-3197.
9. Ross GA, Morris GM, Biggin PC (2012) Rapid and Accurate Prediction and Scoring of Water Molecules in Protein Binding Sites. PLoS ONE 7: e32036.